Understanding Unit Cell Shapes for Molecular Simulations in GROMACS Wizard
Efficiently Export Atom Trajectories Along Defined Paths in SAMSON
Effortlessly Track Molecular Movements with the ‘Follow Atoms’ Animation
Streamline Your Molecular Modeling with a Customizable Interface.
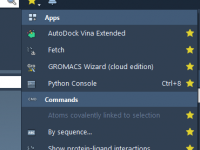
Mastering Custom Molecular Dynamics Parameters in SAMSON’s GROMACS Wizard
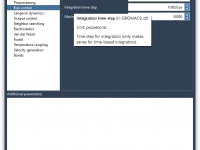
Mastering Keyframe Animation for Moving Atoms in SAMSON
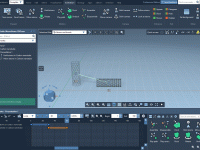
Create Custom HCL Color Palettes in SAMSON
Molecular modelers rely heavily on visualizing structural details clearly and effectively. Whether you’re analyzing protein-ligand interactions or exploring massive molecular assemblies, choosing the right color schemes is critical for interpretation and communication. Sometimes, though, default color palettes might not align…
Tips for Assembling Molecular Fragments in SAMSON Platform
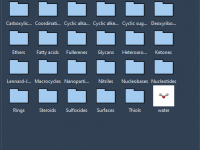
Molecular modelers often face the challenge of efficiently building complex molecular systems. Whether working on proteins, nanoparticles, or small molecules, creating accurate molecular structures quickly and intuitively can be demanding. SAMSON, the integrative molecular design platform, offers a powerful set…