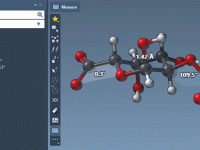
Step-Up Molecular Visualizations with Customizable Visual Presets
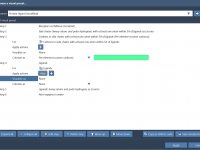
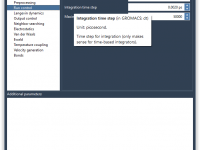
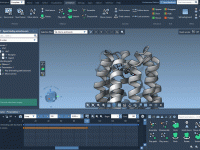
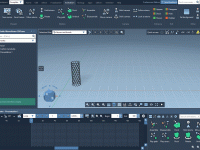
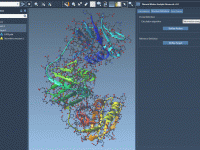
Molecular modelers often grapple with the challenge of efficiently visualizing complex molecular systems. Whether it’s protein-ligand interactions, detailed structural analysis, or preparing impactful presentations, configuring the perfect visualization can be daunting—colors, representations, node selections, and more need careful setup. Enter…