





When working with protein structures, accuracy begins at preparation. One common issue molecular modelers face—especially when dealing with alternate conformations or large multimeric structures—is the need to remove unwanted chains before running simulations, conformational interpolation, or structural comparisons. For instance, if your structure contains multiple chains but your analysis concerns only one, leaving irrelevant chains can lead to misleading results or tool errors.
This step-by-step snippet from the SAMSON documentation focuses on exactly this: how to load and prepare two conformations of a protein, remove unnecessary chains, and clean both structures for robust comparison and interpolation using the ARAP (As-Rigid-As-Possible) technique.
Why This Matters
Many structural datasets from the Protein Data Bank (PDB) contain biological assemblies with multiple chains, ligands, water molecules, and sometimes alternate positions. These elements—while biologically relevant—can interfere with geometric or energetic analyses if not properly handled.
For example, in the ARAP interpolation tutorial, two structures—1DDT and 1MDT—represent different conformations of Diphtheria Toxin. Both contain chains A and B, but the interpolation path is computed only between chain A of each structure. Including chain B would introduce an unwanted level of complexity or even break the interpolation process.
Step-by-Step: Cleaning Protein Structures
- In the SAMSON interface, go to Home > Fetch.
- Fetch
1DDTand1MDTwith either PDB or mmCIF format. - Click Load for each structure. They will appear in your Document view.
Now that you’ve got the structures loaded, here’s what to do next:
- Expand
1MDTin the Document view. - Locate and select chain
B. - Delete it using the Del key or the Erase tool in the popup context menu.
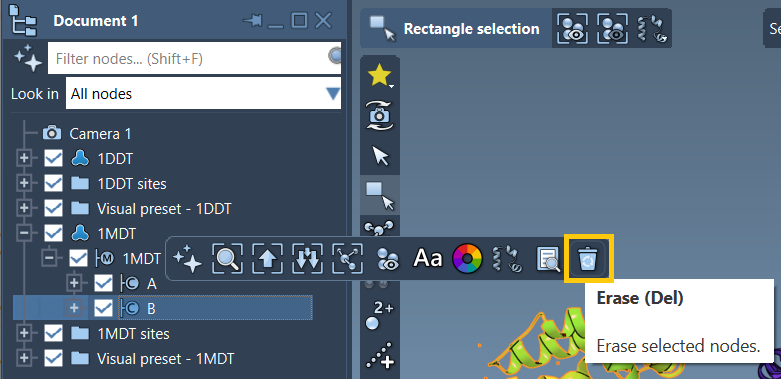
This ensures that both structures (1DDT and 1MDT) now contain only chain A. This is crucial for generating a valid ARAP interpolation path.
Structure Clean-up
Before interpolating, it’s good practice to remove water molecules, ligands, ions, and alternate positions that may affect downstream applications.
To do that, simply run Home > Prepare. This command cleans the structures in one click. If you need more detailed guidance, the Protein Preparation & Validation page explains these steps in-depth.
When You Should Do This
Besides ARAP interpolation, cleaning and pruning structures is recommended before tasks like:
- Molecular Dynamics (MD) simulations
- Conformational clustering
- Free energy computations (e.g., umbrella sampling)
- Machine learning-based structural classification
Whether you’re visualizing motions or preparing inputs for more advanced simulations, well-prepared input structures help avoid costly computational errors and improve reproducibility.
To learn more or continue exploring the interpolation workflow, visit the full documentation at this page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can access and download SAMSON at https://www.samson-connect.net.