





Molecular modeling often involves complex calculations and parameter setups, and for many researchers, streamlining these processes is key to productivity. The Universal Force Field (UFF) in SAMSON offers a robust tool for simulating molecular systems, but figuring out how to get started can leave users scratching their heads. In this post, we’ll walk you through one of the most practical aspects of using UFF: setting it up for a molecular simulation.
Why is UFF Important for Molecular Modeling?
UFF is a full periodic table force field developed for molecular mechanics and molecular dynamics simulations. Its advantage lies in its ability to provide automatic recognition of bonds, bond orders, and atom types, allowing users to focus on their scientific tasks rather than manual parameter tuning. Whether you’re working on new materials or studying molecular interactions, UFF can help simplify and enhance your simulations.
Step-by-Step Guide for Configuring UFF
Here’s how you can set up UFF in SAMSON:
- Open your molecular model: Start by loading or creating a molecular system that you want to simulate.
- Add a simulator: Navigate to
Edit > Simulate > Add simulatoror use the shortcutCtrl + Shift + M(orCmd + Shift + Mon macOS). In the simulator window, select Universal Force Field from the list of interaction models. - Select your updater: Choose the state updater to manage your simulation dynamics. For example, the Fast Inertial Relaxation Engine (FIRE) is a popular choice.
- Configure bonds: In the UFF setup window, decide whether to use existing bonds from your model or allow UFF to re-calculate them automatically from atom positions and types. Selecting Use existing bonds retains your current bond configurations.
- Initialize the system: Click OK, and the module will handle important preparatory steps like computing bonds, bond orders, and atom types. If any inconsistencies are detected, SAMSON provides a warning to help you debug issues.
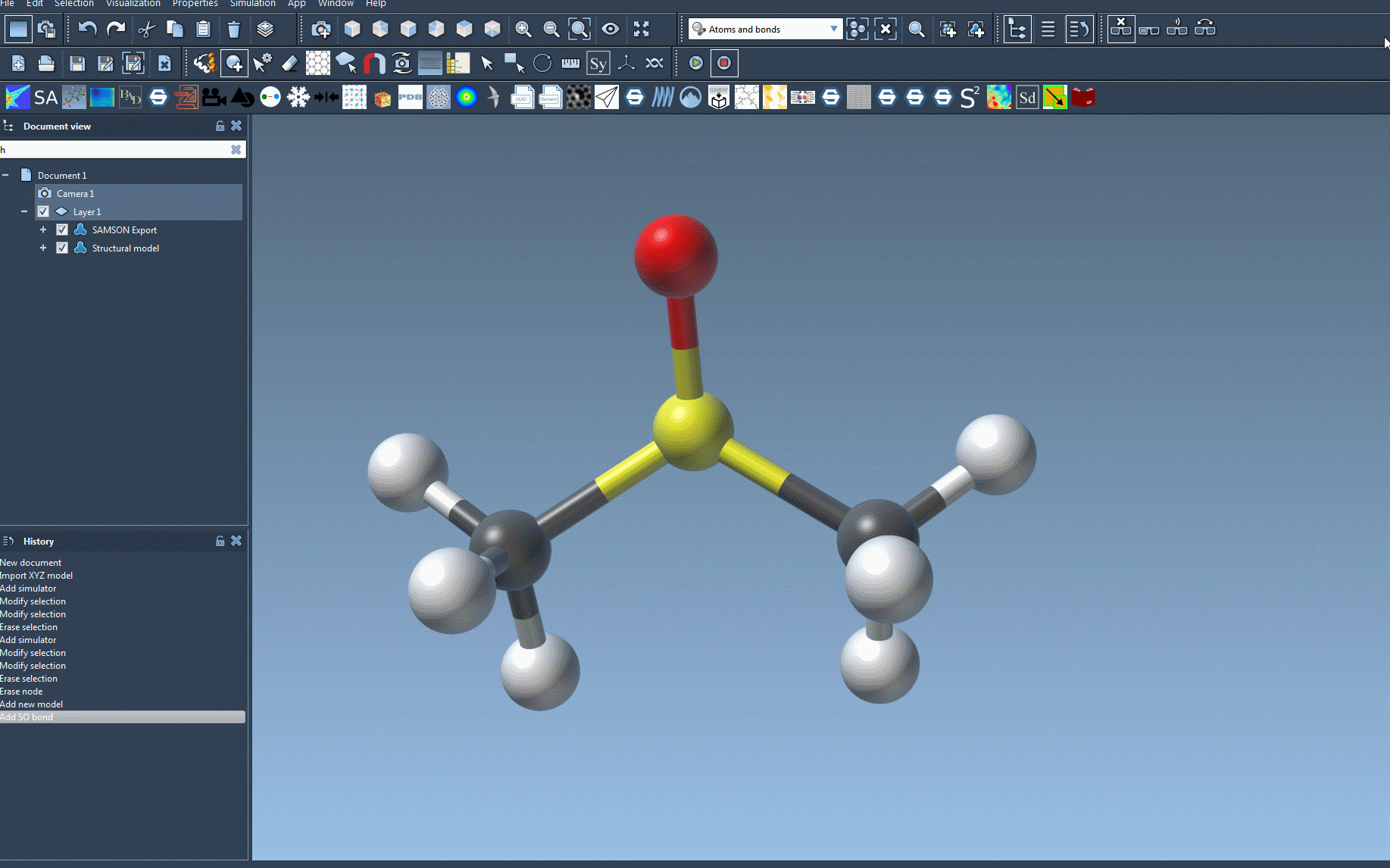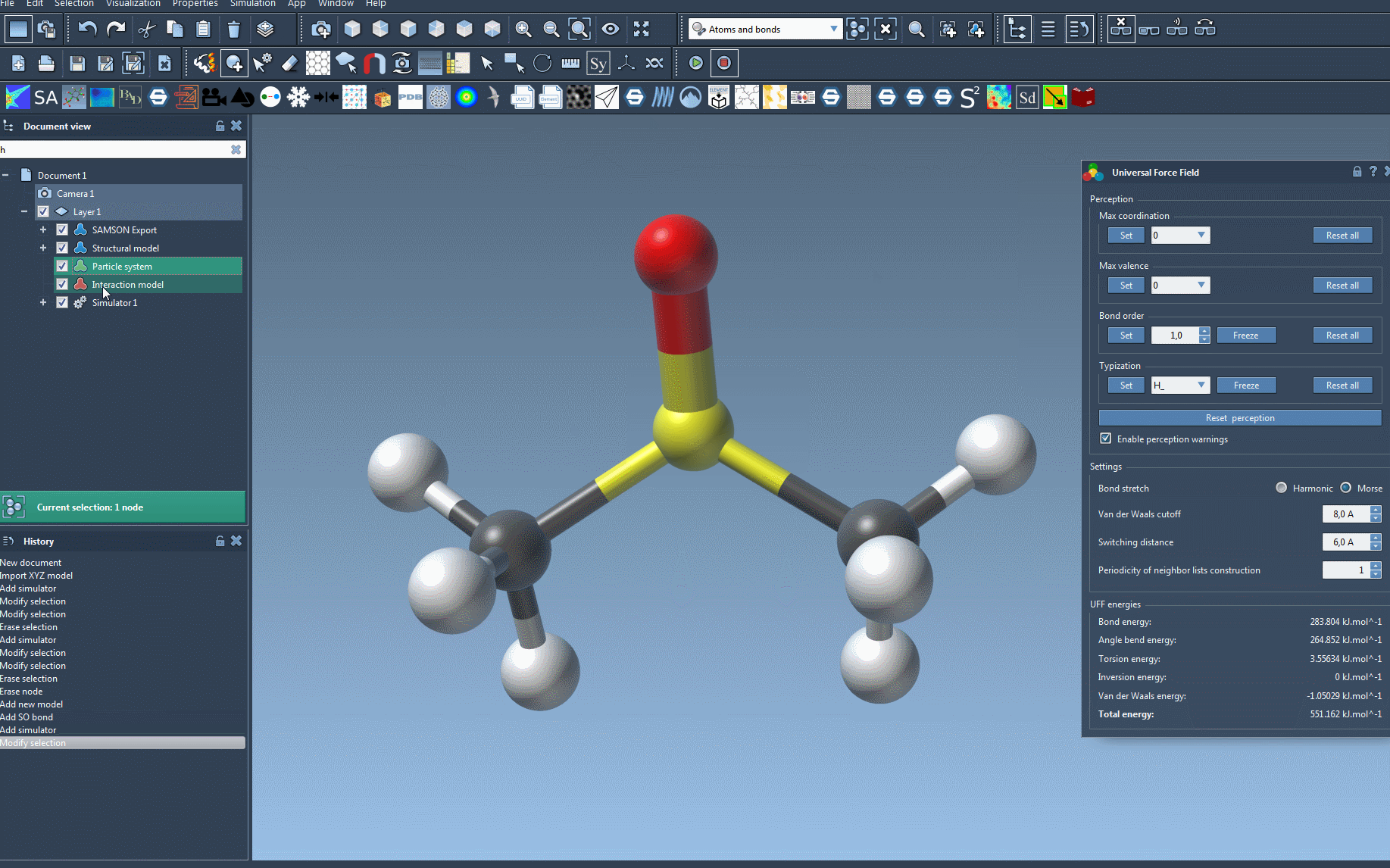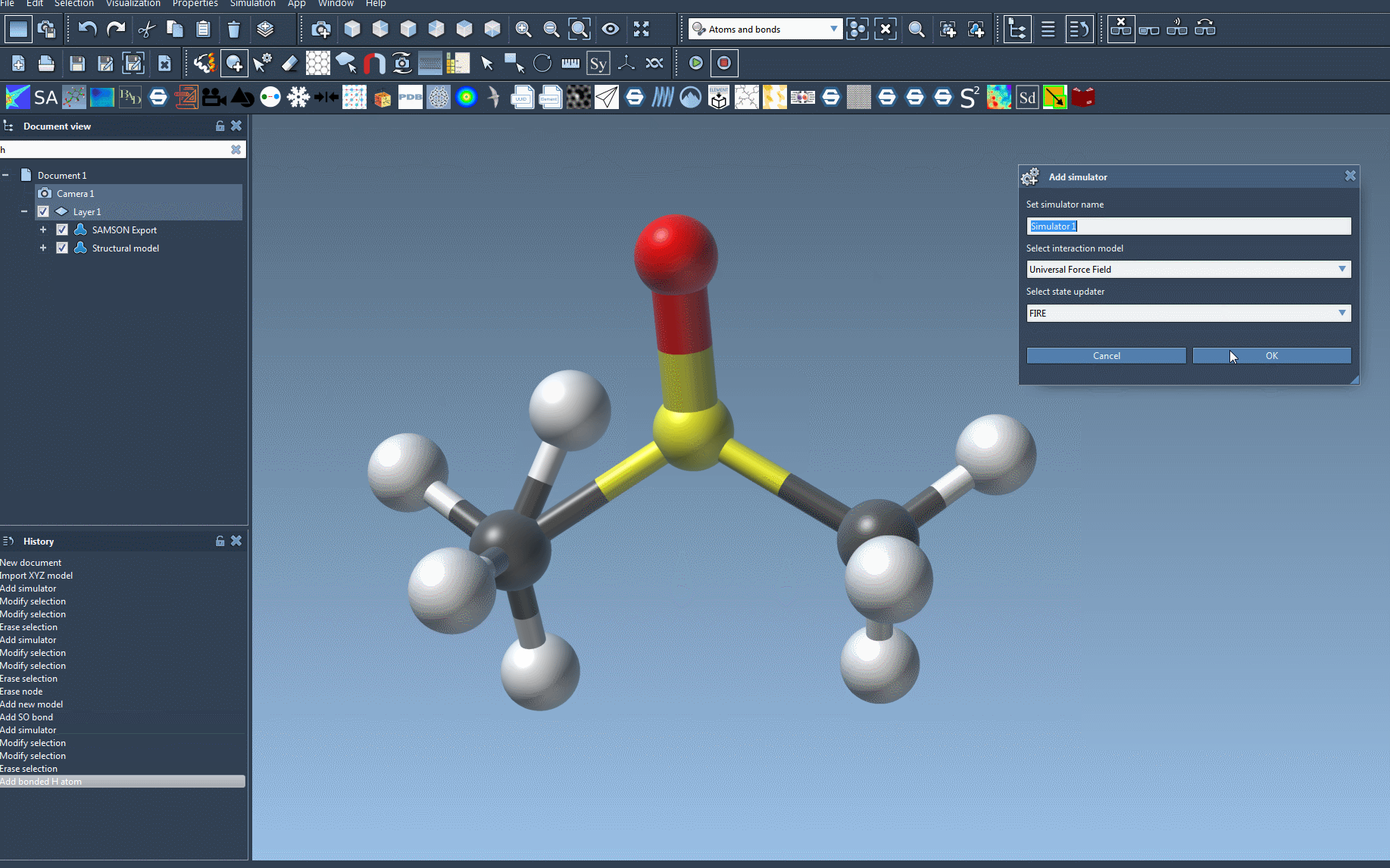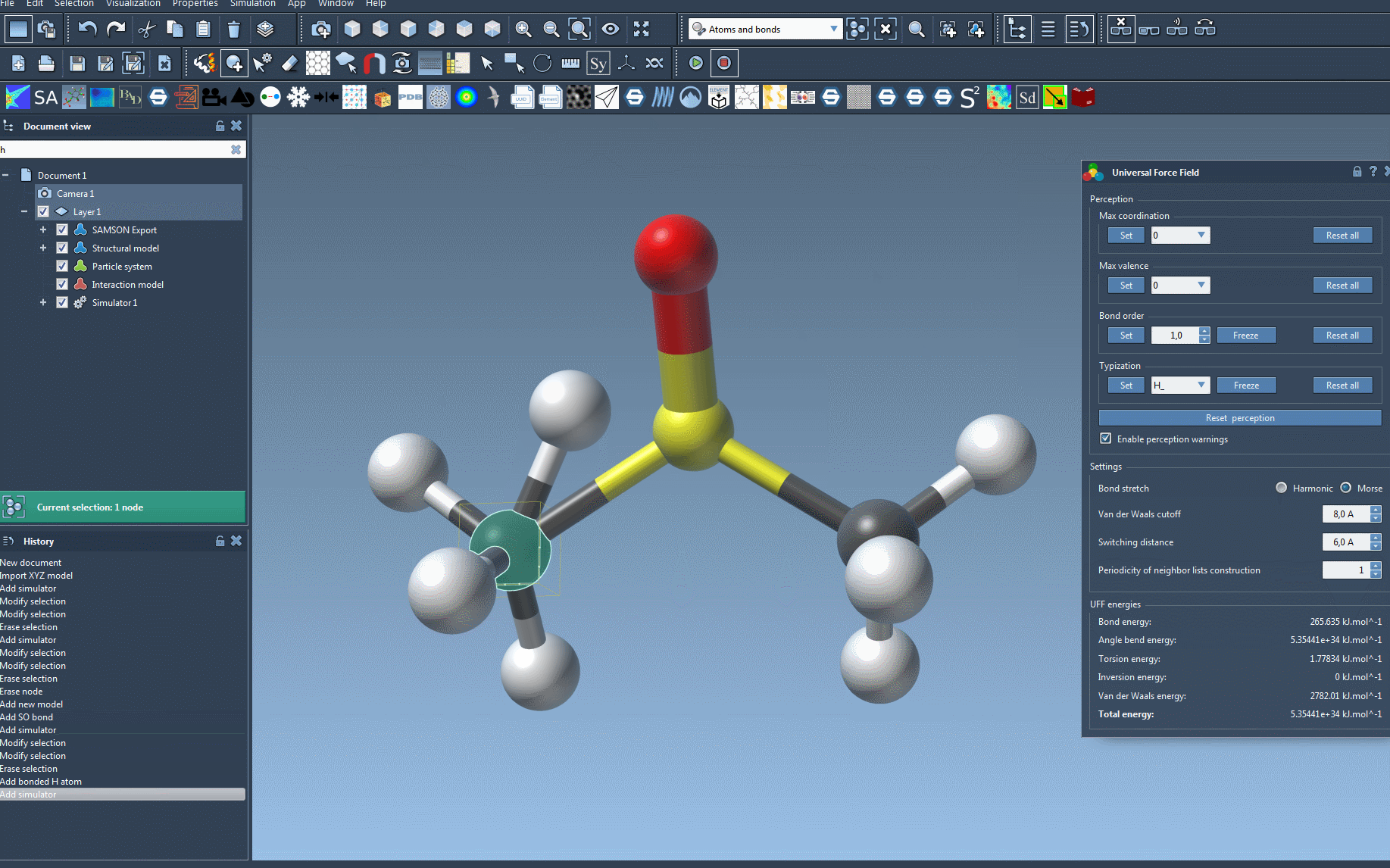
At this point, you will see the UFF parameter window where you can further fine-tune parameters if needed before running your simulation.
Visualizing the Setup Process
To make the setup process easier to visualize, here’s an animation of adding the UFF simulator in SAMSON:
Troubleshooting Tips
- If you don’t see bonds where they should be, verify the accuracy of your molecular structure and consider whether existing bonds or UFF-calculated bonds better fit your simulation goals.
- Check the documentation for default coordination and valence numbers to better understand how UFF interprets atom connectivity. Advanced users can override these settings for more precise control over the perception process.
Next Steps
With the UFF setup complete, you’re ready to start your simulations and dive into molecular insights. UFF is interactive by design, so you can even move atoms during a simulation and see how the system dynamically adjusts. Don’t miss the chance to customize parameters like bond stretch interactions, van der Waals cutoffs, and neighbor list periodicity—these small tweaks can make a big difference.
To learn more about UFF, explore other features, or troubleshoot, visit the full UFF documentation page. Enhance your molecular modeling workflow with SAMSON and take full advantage of this automated yet customizable force field.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at samson-connect.net.