





Transition paths can reveal crucial insights into molecular motions, essential for understanding complex systems like protein-ligand complexes or molecular dynamics in chemical transformations. However, modeling these paths with physical accuracy is often a challenge. Scientists tend to start with an initial rough pathway but struggle to refine it into something more meaningful. This is where the Parallel Nudged Elastic Band (P-NEB) app in SAMSON can become a game-changer.
Why Optimize Transition Paths?
A rough transition path might provide a general sense of molecular trajectories, but it rarely reflects the underlying energy landscape accurately. The P-NEB app integrates the Nudged Elastic Band (NEB) method to address this. This method minimizes the system’s energy along the entire path while ensuring intermediate states (or “images”) remain evenly distributed. The result? A physically meaningful, optimized transition path that aligns better with the system’s energy profile.
How to Get Started
Before you dive into using P-NEB, here are the prerequisites:
- Install the P-NEB app and FIRE state updater.
- Have a rough path or a set of conformations ready. If you don’t already have one, you can use tools like linear interpolation or the Ligand Path Finder Extension to generate an initial pathway.
Core Features of the P-NEB App
The P-NEB app in SAMSON has been designed with flexibility and precision in mind. Before running the optimization, you can adjust key parameters:
- Spring Constant: Keeps neighboring states evenly distributed.
- Number of Loops: Sets how many optimization cycles will be performed.
- Interaction Model: Choose from force fields like “Universal Force Field” for energy computation.
- Optimizer: Use algorithms such as FIRE for optimizing the transition path.
- Parallel Execution: Optimize multiple images simultaneously to save computation time (recommended).
These configurations make the app suitable for both paths and sets of conformations.
Applying P-NEB to a Path
One of the quickest ways to refine a transition path is by directly applying P-NEB to an existing path in SAMSON. Here’s how:
- Step 1: Select your path node in the “Document View.”
- Step 2: Open the P-NEB app (
Home > Apps > All > P-NEB) and configure the core settings. For example, you may set the spring constant to1.00, number of loops to100, and interaction model to “Universal Force Field” as a simple starting point. - Step 3: Click on the Run button. During initialization, select “use existing bonds” if prompted.
As optimization progresses, you can monitor the status bar for real-time updates. Once complete, the refined path will appear in the “Document View.” You may inspect the new path with tools like the “Inspector” or animate it by double-clicking.
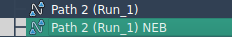
Resolving Set of Conformations
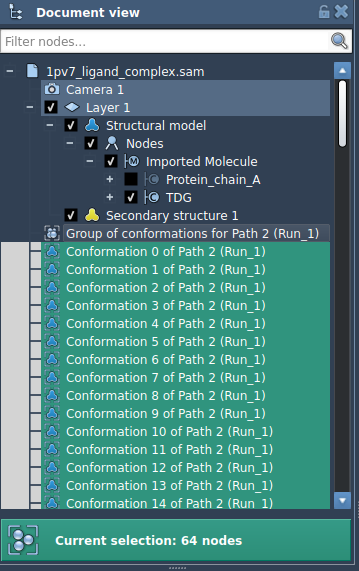
While directly optimizing a path is recommended, you can also apply P-NEB to a sequence of conformations. For the same system, this will typically take longer than working with a path. If you choose to proceed this route, you can streamline the initial setup by combining selected conformations into a path (via Conformation > Create path from conformations).
Once optimized, you’ll see a new set of adjusted conformations in the “Document View,” which you can double-click to modify atomic positions in SAMSON’s interface.
Take Your Research Further
The P-NEB app isn’t just about refining paths—it’s about setting up trajectories that lead to deeper molecular insights. Whether you’re exporting atom trajectories or preparing for free energy workflow studies, the tools in SAMSON and its extensions provide robust support for innovators in molecular modeling.
For detailed technical steps and related guidance, visit the official documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON at https://www.samson-connect.net.