





Periodic boundary conditions are essential for simulating large molecular systems in a computationally efficient way. However, for many molecular modelers, the nuances of these boundaries can raise questions. What do they mean for your simulations? How can you ensure your system is set up correctly? Let’s dive into the topic using the GROMACS Wizard, an extension available in SAMSON, to clarify these crucial aspects of molecular modeling.
The Basics: What Are Periodic Boundary Conditions?
Imagine you’re simulating a solute in a solvent, but instead of having to model an infinitely large amount of solvent, periodic boundary conditions help by replicating a small, finite unit of the system in all directions. This makes your molecular dynamics simulation computationally feasible.
One key idea is the minimum image convention. In this approach, each particle sees and interacts with only the nearest copy of another particle, ensuring computational efficiency without compromising simulation accuracy. For most systems, this requires a proper distance between the solute and the boundaries of the simulation box.
Choosing the Right Unit Cell for Your Simulation
The unit cell is the box containing your system that gets replicated. Different unit cell shapes can make a big difference in how efficiently you model your system. The GROMACS Wizard supports several unit cell shapes:
| Unit Cell Shape | Visualization |
|---|---|
| Cubic |  |
| Orthorhombic |  |
| Triclinic |  |
| Rhombic Dodecahedron |  |
| Truncated Octahedron |  |
Why Shape Matters
Choosing the optimal unit cell shape depends on your system. For instance:
- The rhombic dodecahedron and truncated octahedron are closer to spherical in shape, reducing the number of solvent molecules required for simulations of approximately spherical macromolecules like proteins. This saves both computational time and resources.
- The rhombic dodecahedron is particularly efficient, with a volume that is about 71% of a cube of the same image distance. This means fewer solvent molecules need to be simulated for the same minimum image convention distance.
On the other hand, cubic or orthorhombic boxes may be more natural choices for less spherical systems. Using the GROMACS Wizard, you can easily select and visualize the most appropriate unit cell for your molecular simulations.
Setting Up Your Box
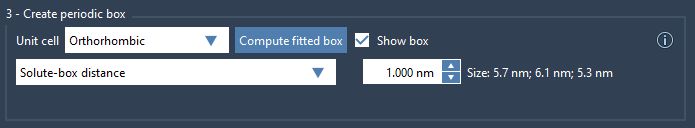
The GROMACS Wizard provides two options for defining your simulation box:
- Box lengths: This allows you to specify exact dimensions for the box. It’s ideal when you want consistent box sizes across a batch of simulations but might require you to tweak the size later to maintain the minimum image convention.
- Solute-box distance: By specifying the distance between the solute and the box, you ensure that the solute never interacts with its own periodic images. A recommended value is 1.0 nm, which translates to a 2.0 nm gap between periodic images.
An example of using this feature can be seen below:
Final Notes
Using the GROMACS Wizard effectively can save you hours of tweaking simulations and interpreting results. Beyond just being a modeling tool, it ensures your simulation adheres to the fundamental principles of periodic boundary conditions. For further details, you can explore the official documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON here.