





For molecular modelers, identifying accurate transition paths in complex systems is often a critical task. Whether you’re studying protein-ligand interactions or conformational changes in macromolecules, refining initial guesses into meaningful pathways can be a daunting challenge. The Parallel Nudged Elastic Band (P-NEB) method in SAMSON is designed to address this pain point by optimizing your rough paths or sets of conformations into physically meaningful transitions with ease.
Why Transition Path Optimization Matters
At the heart of computational chemistry and molecular modeling lies the quest to understand how systems evolve from one state to another. Transition paths, which describe these processes, often connect local energy minima in a potential energy landscape. However, raw conformations or straight-line interpolations may not accurately capture intermediate structures or provide realistic energetic profiles.
The P-NEB approach allows you to refine these transition paths into scientifically robust trajectories optimized against force-field-based interaction models while maintaining even distribution of conformations using spring forces.
How the P-NEB App Simplifies Pathway Refinement
Once you have a preliminary candidate pathway—be it a rough trajectory or a set of conformations—the P-NEB app in SAMSON enables the optimization process through a user-friendly interface. Here’s how it works:
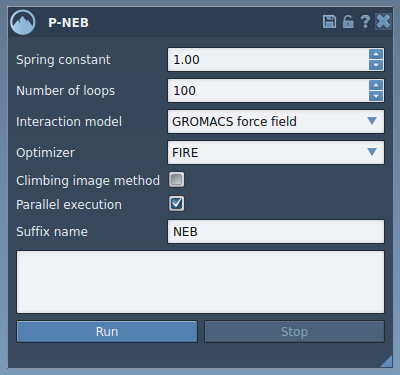
The app lets you:
- Define a spring constant to maintain equal spacing between intermediate conformations.
- Set the number of optimization loops for refining the transition path.
- Select the interaction model (e.g., the Universal Force Field) to calculate energies and forces.
- Apply a climbing image method to home in on high-energy saddle points (optional).
- Leverage parallel execution capabilities to handle complex systems efficiently.
For example, you might take an initial ligand unbinding pathway generated by the Ligand Path Finder in SAMSON, then refine it with P-NEB to obtain a highly accurate model of the ligand’s trajectory.
Step-by-Step: Optimizing a Path
To use the P-NEB app, start by opening SAMSON and ensuring you have the necessary extensions installed, including the P-NEB extension. Once ready, follow these steps:
- Select your initial path in the Document view.
- Open the P-NEB app via Home > Apps > All > P-NEB.
- Adjust the parameters as needed, such as setting the spring constant (e.g., 1.00), selecting the FIRE optimizer, enabling parallel execution, and leaving the climbing image unchecked for your first pass.
- Click Run. During computation, you can monitor progress directly in the status bar:

Once complete, a new optimized path will appear in the Document view:
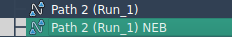
You can double-click this path to animate transitions or examine properties using the Inspector tool.
Tips for Working with Conformations
The P-NEB app can also process a set of conformations directly. However, since direct paths tend to be computationally more efficient, it’s recommended to combine conformations into a single path before optimization. To do this, select multiple conformations in the Document view, then right-click and use the option Conformation > Create path from conformations.
Working Towards Better Molecular Insights
With the P-NEB method in SAMSON, the task of refining transition paths becomes approachable and efficient, saving molecular modelers time while delivering results rooted in physical rigor. This tool is particularly valuable for those studying complex molecular systems and aiming to understand key processes like ligand unbinding or protein folding with greater accuracy.
To dive deeper into optimizing transition paths using the P-NEB app, visit the official documentation page: https://documentation.samson-connect.net/tutorials/pneb/optimize-transition-paths-with-parallel-nudged-elastic-band/.
SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON at https://www.samson-connect.net.