Author: OneAngstrom
Understanding Visual Model Attributes for Streamlined Molecular Modeling in SAMSON
For molecular modelers, managing the appearance and behavior of visual models is essential when working with large, complex systems. Whether you are clarifying a visualization for publication, streamlining your workflow, or creating detailed models for analysis, controlling visual attributes efficiently…
Mastering Side Chain Attributes in Molecular Modeling
Making SAMSON Work for You: Interface Customization Tips for Molecular Modelers
Efficiently Handling Ligand Flexibility in AutoDock Vina Extended with SAMSON
Exploring Adenita’s Interface: A Functional Walkthrough
DNA nanotechnology presents fascinating opportunities for scientific advancement, but effectively designing nanostructures requires powerful tools with intuitive interfaces. Enter Adenita, a SAMSON Extension created to optimize DNA nanostructure design workflows. In this post, we’ll explore how Adenita’s interface is structured…
Effortlessly Track Protein Secondary Structures Over Time.
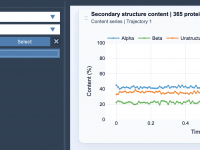
Visualizing Contacts for Molecular Interactions
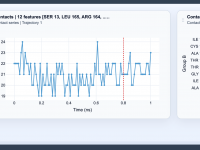
Molecular modelers frequently grapple with the challenge of understanding and monitoring interactions between molecules, atoms, or structural features over time. Whether you’re investigating intricate binding events, tracking the packing of domains, or analyzing key molecular interfaces, clarity on how contacts…