Author: OneAngstrom
Mastering Animation Attributes in SAMSON’s NSL
Streamlining Molecular Design: Understanding SAMSON Importers
Stay Focused: Using ‘Look at Atoms’ Animation for Molecular Modeling
Master Your Molecular Modeling Workflows with SAMSON’s Interactive Tutorials
Streamlining NVT Equilibration with the GROMACS Wizard in SAMSON.
For molecular modelers, setting up simulations to ensure temperature stabilization can often feel daunting. A common challenge is running NVT equilibration (constant Number of particles, Volume, and Temperature) effectively, especially with tools that aren’t intuitive to use. Enter SAMSON’s GROMACS…
Unpacking Local Packing with RDF Analysis in SAMSON
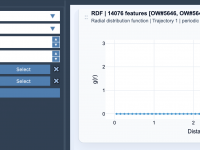
When studying the molecular world, understanding how components are distributed around each other is essential, whether you’re delving into solvation structures, evaluating drug interactions, or analyzing local packing. This is where Radial Distribution Function (RDF) analysis becomes valuable. In this…
Understanding and Registering Monomer Sequences in SAMSON’s Polymer Builder
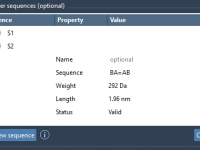
For molecular modelers and researchers working on custom polymer design, one of the biggest challenges is handling sequences effectively when building large, complex structures. Whether you’re optimizing for specific material properties, creating biopolymers, or simulating new synthetic chains, accurately registering…