Author: OneAngstrom
Simplify Collaboration: A Guide to Sharing and Managing Documents on SAMSON Connect
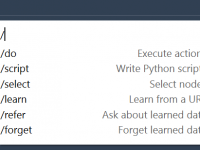
Unlocking Efficiency in Molecular Modeling with SAMSON AI Commands
Effortless Vertical Moves with the Pedestal Camera Animation
Master Molecular Workflows with SAMSON’s Interactive Tutorials
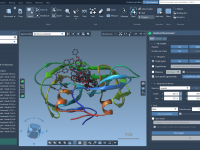
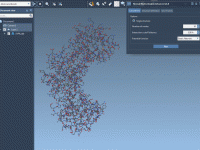
Exploring the Shape Parameter for Molecular Design Insights
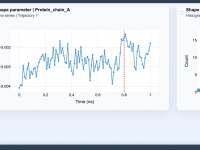
For molecular modelers, the ability to effectively analyze and interpret the shapes of molecular trajectories is crucial, especially when studying complex biochemical processes or designing new molecules. However, traditional shape descriptors like asphericity or radius of gyration might not always…
Highlight and Replace Patterns in Molecules Using SMARTS Codes.
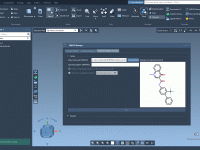
For molecular modelers, efficiently analyzing and generating variations of molecular structures can often be a time-intensive task. Highlighting specific parts of a molecule to explore substitutions or modifications can dramatically accelerate workflows for tasks such as structure optimization or molecular…
Loading Your First Molecular Structure in SAMSON
Mastering Custom Index Groups in GROMACS Wizard
If you’re a molecular modeler working with GROMACS, chances are you’ve encountered scenarios where the default index groups provided during simulations are insufficient for your project. Whether you’re looking to define pull groups, isolate specific residues, or streamline post-simulation analyses,…