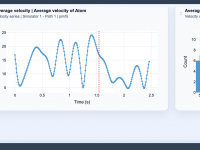
Author: OneAngstrom
Streamlining Molecular Visualization with Visual Presets in SAMSON
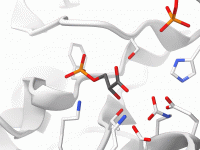
Molecular modelers often need to create visually compelling and informative representations of complex molecular systems. This can involve customizing multiple parameters like visual representations and color schemes—a time-intensive process. Luckily, SAMSON offers a solution: Visual Presets. They allow users to…
Mastering the Universal Force Field Setup in SAMSON
Mastering Interactive Carbon Nanotube Creation in SAMSON

For molecular modelers, nanotechnology enthusiasts, or materials scientists, customizable carbon nanotube (CNT) models are a must-have. But creating these complex structures can be a frustrating challenge if you’re working without intuitive tools. What if you could build single-walled or multi-walled…
Ensuring Compatibility for Your SAMSON Extensions
Boost Your Molecular Modeling Workflow with SAMSON Extensions

Understanding Residue Polarity for Molecular Modeling
Mastering Sequence Selection Across Views in SAMSON
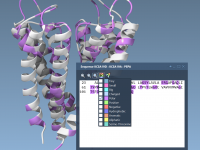
Molecular modelers often face the challenge of managing and selecting specific residues in complex molecular structures. Whether you’re focusing on a particular chain, analyzing biophysical properties, or preparing data for workflows, accurate and synchronized selection across multiple views is crucial.…