





Author: OneAngstrom
Understanding and Manipulating Crystals: A Practical Guide for Molecular Modelers
For molecular modelers, accurately manipulating and understanding crystal properties is a critical task that has significant implications for various material science applications, such as nanoengineering, molecular design, and structural analysis. SAMSON’s Crystal Creator Extension provides powerful tools to not only…
Streamline Molecular Visualizations with the Pulse Animation in SAMSON
Streamlining Molecular Simulations with UFF Setup in SAMSON
Mastering the Disappear Animation for Molecular Models
Mastering Custom Molecular Dynamics Parameters in SAMSON’s GROMACS Wizard
Crafting a Public Profile for Collaboration on SAMSON Connect
For molecular modelers, building connections and sharing insights efficiently can be challenging. Crafting an accessible and well-organized public profile can help improve visibility and collaboration within the scientific community. SAMSON Connect makes this process intuitive, allowing you to share information…
A Practical Guide to Refining Transition Paths with P-NEB in SAMSON
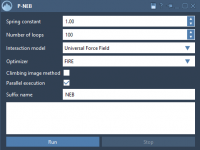
Molecular modeling often involves studying complex energy landscapes to understand transition pathways between states. Whether you are investigating ligand unbinding, protein conformational changes, or reaction mechanisms, obtaining accurate and physically meaningful transition paths can be challenging, especially if your initial…