Author: OneAngstrom
Enhance Your Modeling with SAMSON’s Dark Mode
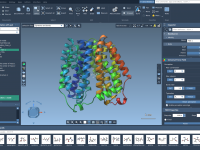
Molecular modelers often spend extended periods analyzing molecular structures, tweaking settings, and running calculations. While the default interface settings in software platforms are often functional, they may not always provide the most comfortable experience—especially during long work sessions or in…
Making Protein Conformation Analysis Interactive with the Ramachandran Plot App

Streamline Molecular Docking with the Dock Animation in SAMSON
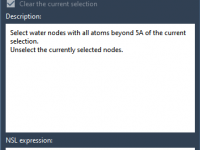
Refining Your System: Removing Crystal Waters Outside the Active Site.
Effortless Visualization with Pulse Animation in SAMSON
Effortlessly Manage Molecular Modeling Jobs in the Cloud

Molecular modeling often involves computationally intensive tasks, like protein structure prediction or molecular dynamics simulations. While essential, managing these workloads can be challenging, especially when operating at scale or collaborating across teams. This is where the Job manager in SAMSON…