Author: OneAngstrom
Exploring Defects in Diamond Structures with SAMSON.
Mastering Chain Attributes in SAMSON’s Node Specification Language
Molecular modelers often face challenges when working with complex systems, especially when navigating and specifying chains within their structures. SAMSON’s Node Specification Language (NSL) offers a powerful toolset for detailing and filtering chain-level attributes effectively. In this blog post, we’ll…
Streamlining Molecular Visualization with Visual Presets in SAMSON
Molecular modeling can often involve managing complex systems with various components that require distinct visualization styles. Switching back and forth between different representations can feel like a daunting task, especially when working with large molecular systems or preparing visualizations for…
How to Customize Atom Typization in Universal Force Field Simulations
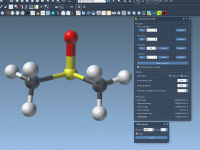
Streamline Molecular Modeling with Easy Universal Force Field (UFF) Setup
Effortlessly Running GROMACS in the Cloud with SAMSON
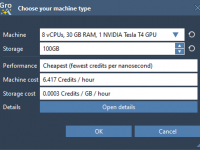
Simplify Molecular Modeling with Chain Attributes in SAMSON
Aligning Protein Sequences: A Workflow for Molecular Modelers
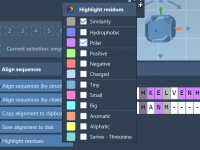
For molecular modelers, understanding the nuances of protein functions often hinges on comparing sequences and structures. Whether you’re investigating conserved residues or studying conformational changes, protein alignment is a crucial step. SAMSON’s Protein Aligner offers an intuitive and efficient workflow…