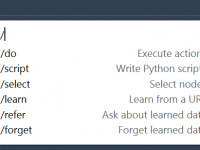
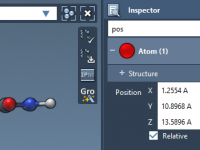
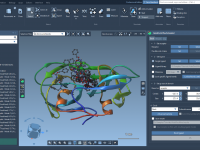
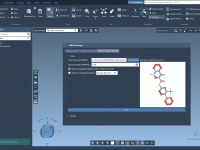
Simplifying Protein Motion with Sampling Boxes.

Molecular modelers often face the challenge of efficiently simulating complex protein motion while maintaining control over computational resources. This balance can be especially tricky when determining transition pathways between two conformations of a protein. Defining the sampling box—a key feature…