





Author: OneAngstrom
Simplifying Energy Minimization for Molecular Systems in SAMSON’s GROMACS Wizard.
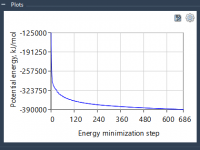
Solving Steric Clashes with GROMACS Wizard’s Energy Minimization
Simplify Your Molecular Modeling with Chain Attributes in SAMSON
Streamlining Atom Trajectory Export for Molecular Modeling
Tracking atom trajectories through molecular pathways forms the backbone of simulations in molecular modeling. However, many researchers face challenges when trying to extract atomic coordinates along defined paths for advanced analyses like reaction coordinate studies or free energy calculations. If…
Streamlining DNA Nanostructure Design with Adenita.
Simplify Your Transition Path Optimization with the P-NEB App
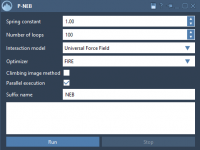
Finding transition paths between molecular states can often be a challenging task for molecular modelers. Whether you’re working on uncovering reaction mechanisms or exploring conformational changes, capturing these pathways accurately and efficiently is essential. This is where the Parallel Nudged…
Mastering Molecular Selection with Node Specification Language (NSL)
Getting Started with the /do Command in SAMSON AI
Mastering the Dolly Camera: Enhancing Molecular Animations
For molecular modelers, effectively presenting dynamic systems can be a crucial part of their workflow. Whether you’re showcasing molecular interactions, structural changes, or effect-driven cinematic storytelling, camera control in animations is key to immersive visualization. One particularly versatile tool for…