Author: OneAngstrom
Streamlining Molecular Modeling with Interactive Tutorials
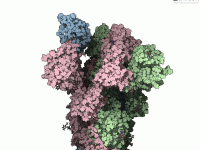
Making Your Molecular Data Visually Intuitive in SAMSON.

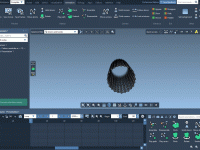
Effortlessly Create Molecular Patterns in SAMSON
Building complex molecular patterns can be a daunting task for molecular modelers. Whether it’s constructing nanotubes, designing curved molecular shapes, or arranging structures in intricate patterns, the process is often time-consuming and prone to errors. Fortunately, SAMSON’s Pattern Building Editors…
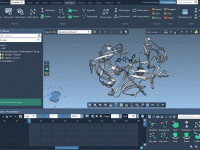
Disassemble Molecular Structures with Ease in SAMSON
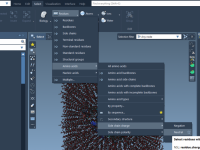
Unlocking Node Visibility in SAMSON: A Guide for Molecular Modelers
For molecular modelers working with complex structures, managing visibility settings for nodes can be a game-changer in streamlining workflows and efficiently analyzing molecular data. SAMSON, the integrative molecular design platform, offers detailed control over node visibility, but there’s more to…