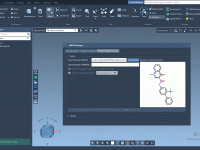
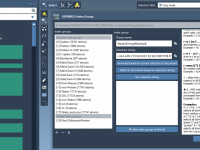
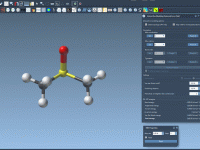
Streamline Protein-Ligand Modeling with 2D Interaction Diagrams
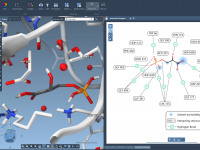
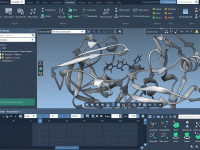
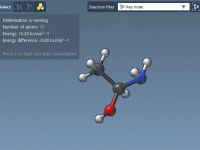
For researchers and molecular modelers working with protein-ligand interactions, visualizing and exploring molecular interactions effectively is essential yet often challenging. The SAMSON Interaction Designer offers a user-friendly way to bridge the gap between 3D and 2D molecular modeling, enabling researchers…