Author: OneAngstrom
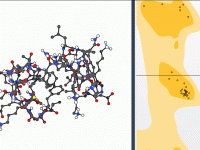
Making Ramachandran Plots Interactive for Protein Modeling
Simplify Molecular Modeling: How to Manage and Install SAMSON Extensions
Making SAMSON Icons Larger on High DPI Monitors
Understanding Render Preset Attributes in SAMSON
For molecular modelers, visualizing molecular systems effectively is often a critical part of their work. The appearance of molecules on screen can significantly influence analysis, presentations, and communication of results. SAMSON’s render preset attributes in the Node Specification Language (NSL)…
Understanding Render Preset Attributes in SAMSON’s NSL
Preparing Your Linux System for SAMSON
Streamline Your Molecule Setup with the FIRE Minimizer

Geometry optimization is a crucial step in molecular modeling. It helps ensure stable and physically realistic molecular structures, setting the stage for reliable simulations and accurate molecular design workflows. However, optimizing molecular geometries can be a time-consuming challenge, especially when…
Understanding Label Attributes in SAMSON’s Node Specification Language
Molecular modelers often face challenges when working with large datasets, especially when it comes to categorizing and managing information about specific molecular nodes. SAMSON’s Node Specification Language (NSL) provides targeted solutions to these challenges through its label attribute space. In…