





Category: Uncategorized
Refining Input Selection for Seamless GROMACS MD Simulations.
Enhancing Molecular Modeling with the ‘Reveal Atoms’ Animation in SAMSON
Discover the Power of Extending SAMSON with Extensions
How to Register Custom Monomers in SAMSON’s Polymer Builder
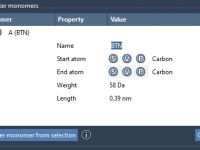
For molecular modelers, flexibility and precision in constructing custom polymers are critical. Whether you are designing synthetic chains for material science or tailoring biopolymers for simulation, the ability to register and manipulate base units (monomers) efficiently can significantly speed up…
A Molecular Modeler’s Guide to Exporting with SAMSON
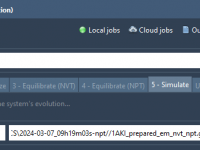
Understanding and Neutralizing Molecular Systems in SAMSON for GROMACS Simulations
Simplify Vertical Camera Movements with the Pedestal Camera Animation
Understanding Residue Secondary Structures in SAMSON
For molecular modelers, analyzing and categorizing the secondary structure of residues—such as distinguishing between alpha helices, beta strands, and unstructured regions—can be essential for insights into structural biology and biochemistry. SAMSON’s Node Specification Language (NSL) offers an intuitive and efficient…
Quickly Filter Specific Atoms in Molecular Models Using NSL
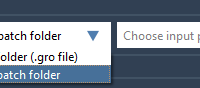
Refining Input Selection for Seamless GROMACS MD Simulations.
Enhancing Molecular Modeling with the ‘Reveal Atoms’ Animation in SAMSON
Discover the Power of Extending SAMSON with Extensions

How to Register Custom Monomers in SAMSON’s Polymer Builder
For molecular modelers, flexibility and precision in constructing custom polymers are critical. Whether you are designing synthetic chains for material science or tailoring biopolymers for simulation, the ability to register and manipulate base units (monomers) efficiently can significantly speed up…
A Molecular Modeler’s Guide to Exporting with SAMSON
Understanding and Neutralizing Molecular Systems in SAMSON for GROMACS Simulations
Simplify Vertical Camera Movements with the Pedestal Camera Animation
Understanding Residue Secondary Structures in SAMSON
For molecular modelers, analyzing and categorizing the secondary structure of residues—such as distinguishing between alpha helices, beta strands, and unstructured regions—can be essential for insights into structural biology and biochemistry. SAMSON’s Node Specification Language (NSL) offers an intuitive and efficient…