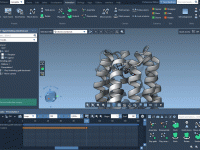
Smooth Molecular Motion: A Practical Guide to Atom Animation in SAMSON
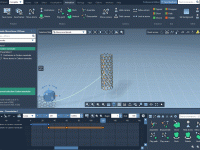
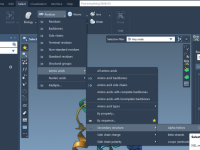
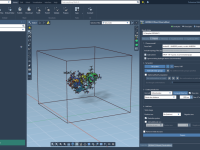
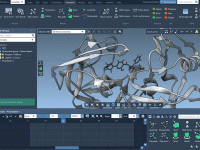
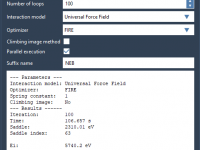
When preparing scientific presentations or molecular animations, one common frustration faced by structural biologists and molecular modelers is presenting complex atomic transformations in a clear, smooth, and repeatable way. Often, the motion of molecules is illustrated frame-by-frame, requiring tedious positional…