





Category: Uncategorized
Making Sense of Molecular Models: How to Filter Backbones by Atom Counts in SAMSON
Interactive φ/ψ Editing: A Faster Way to Tweak Protein Backbone Conformations
Protein structure modeling often involves spotting and correcting outlier conformations in the backbone. These problematic residues can affect simulation stability, energy minimization, or interpretation of function. Fixing them typically means switching between raw coordinate editing or laborious script-based correction. But…
Avoiding common selection mistakes with presentation nodes in SAMSON
Filtering Side Chains by Atomic Composition in SAMSON
Avoid Jumpy Movements When Animating Molecular Systems
When presenting complex molecular transformations or simulations, one of the most frustrating problems molecular modelers face is jittery or discontinuous motion. Whether you’re creating animations to communicate results or to better understand molecular processes, the key to clear, compelling molecular…
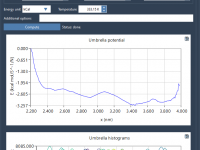
A Quick Path to PMF Profiles with WHAM in GROMACS Wizard
When Bonds Break: Handling Topology Changes in Interactive Molecular Modeling
Make Visual Selection Easier in SAMSON with Render Preset Attributes
Simulating Multiple Conformations at Once: A Time-Saving Workflow for Molecular Modelers
One of the recurring challenges in molecular modeling is evaluating how different conformations of a single molecular system behave under identical simulation conditions. Whether you’re performing Umbrella Sampling or exploring conformational variability, setting up individual simulations for each structure can…
Making Sense of Molecular Models: How to Filter Backbones by Atom Counts in SAMSON
Interactive φ/ψ Editing: A Faster Way to Tweak Protein Backbone Conformations
Protein structure modeling often involves spotting and correcting outlier conformations in the backbone. These problematic residues can affect simulation stability, energy minimization, or interpretation of function. Fixing them typically means switching between raw coordinate editing or laborious script-based correction. But…
Avoiding common selection mistakes with presentation nodes in SAMSON
Filtering Side Chains by Atomic Composition in SAMSON
Avoid Jumpy Movements When Animating Molecular Systems
When presenting complex molecular transformations or simulations, one of the most frustrating problems molecular modelers face is jittery or discontinuous motion. Whether you’re creating animations to communicate results or to better understand molecular processes, the key to clear, compelling molecular…
A Quick Path to PMF Profiles with WHAM in GROMACS Wizard
When Bonds Break: Handling Topology Changes in Interactive Molecular Modeling
Make Visual Selection Easier in SAMSON with Render Preset Attributes
Simulating Multiple Conformations at Once: A Time-Saving Workflow for Molecular Modelers
One of the recurring challenges in molecular modeling is evaluating how different conformations of a single molecular system behave under identical simulation conditions. Whether you’re performing Umbrella Sampling or exploring conformational variability, setting up individual simulations for each structure can…