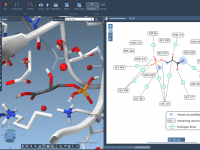
If you’ve ever tried to interpret complex molecular interactions from a dense 3D structure alone, you know how quickly things can become visually overwhelming. Identifying how a ligand interacts with a protein can require constant rotations, manual measurements, and lots…
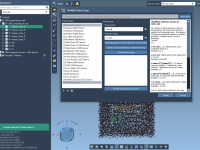
If you’ve ever built or analyzed a large molecular model in SAMSON, you know how quickly complexity can escalate. Hundreds or thousands of groups—sometimes with overlapping naming schemes or similar visual features—can make it tricky to select precisely what you…
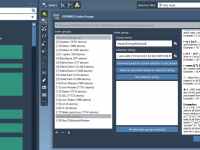
One of the common challenges in setting up center-of-mass (COM) pulling simulations using GROMACS is defining the correct groups of atoms when default index groups are not sufficient. If you’re dealing with multi-chain molecular systems and want to precisely control…
Presenting molecular structures effectively is often as important as building them. Whether you’re preparing a lecture, visual abstract, or research video, storytelling through molecule animations can engage your audience in ways static images can’t. But one recurring challenge many molecular…
When working on molecular systems, it’s common to juggle multiple files and structures at once. You might be preparing different configurations, comparing docking poses, or visualizing simulation results side by side. In such scenarios, quickly identifying which nodes are currently…
When communicating complex molecular models, visual clarity is essential. However, modelers often face a recurring challenge: how can you guide an audience’s attention to specific components of a system without completely deleting or permanently altering the structure? This is especially…
One of the recurring challenges when setting up molecular dynamics (MD) simulations with GROMACS is defining custom index groups. These groups provide more control during analysis and simulation by letting you specify exactly which atoms or residues you want to…
Designing and modeling DNA origami structures involves multiple tools and file formats, which can make managing and sharing your designs a bit challenging. If you work in DNA nanotechnology and are looking for a more integrated approach, SAMSON may help…
When presenting complex molecular systems, timing and clarity matter. Whether you’re giving a seminar, teaching molecular modeling concepts, or creating video tutorials, being able to pause and explain specific moments in your molecular animations is crucial. This is where SAMSON’s…
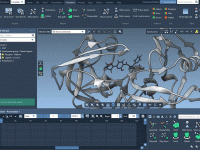
When you’re trying to optimize the properties of a lead compound, exploring small chemical changes at specific sites can be a powerful strategy. Medicinal chemists often want to tweak one part of a molecule and see how it affects activity…