When collaborating across institutions and disciplines, visibility and clarity of roles make a real difference. As a molecular modeler or computational scientist, you’ve probably shared files across cloud drives and sent endless emails introducing yourself and your expertise. But what…
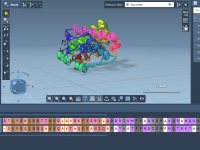
When working with complex molecular systems in SAMSON, switching focus between different molecular components—like ligands, protein domains, or specific residue types—can become tedious. Typically, every time you want to reselect a region, you might need to dig through the Document…
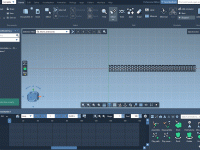
When presenting molecular systems or creating visual walkthroughs in SAMSON, scientists and educators often run into a familiar challenge: how to create smooth, clear animations that allow viewers to follow complex structures horizontally across a scene. While rotating and zooming…
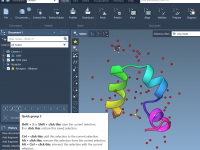
If you’ve ever worked with biomolecular structures, you’ve likely faced the challenge of identifying specific types of residues—such as acidic side chains, positively charged amino acids, or those with specific pKa ranges. These tasks are essential for setting up simulations,…
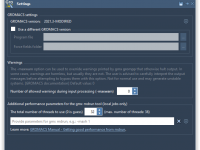
One common frustration among molecular modelers is the unresponsiveness of their computer when running local simulations. You’ve likely experienced it: you launch a simulation, and suddenly everything slows to a crawl. This often happens when computational tools like GROMACS run…
When building or analyzing complex molecular systems, molecular modelers often need fine control over which atoms to keep, manipulate, or hide. Especially in large or periodic structures, identifying a specific spatial region—like a nanodisk from a larger sheet or defining…
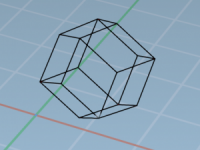
When setting up molecular dynamics simulations, especially using GROMACS, researchers often face a surprisingly impactful decision: what shape should the simulation box be? Choosing the right periodic unit cell shape can significantly reduce computational costs and resource usage, especially when…
In molecular modeling, aligning full protein structures is a common task—especially when comparing proteins from different organisms or assessing mutations. But what happens when you’re only interested in a specific functional domain or a particular secondary structure? This is a…
In collaborative molecular modeling projects, especially in academic and interdisciplinary environments, it is often difficult to keep track of who is doing what. Profiles end up being scattered across platforms, documents shared by email or chat, and institutional pages remain…
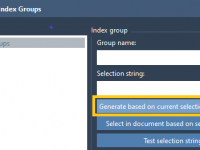
For many molecular modelers, gaining fine control over simulations goes beyond simply setting up a system. Often, it’s just as important to define custom groups of atoms or residues for analysis, restraints, or biasing methods like umbrella sampling. This is…