Category: Uncategorized
Faster Nanotube Building with Pattern Editors: A Step-by-Step Guide
Controlling Molecular Visibility in Time with the Hidden Animation
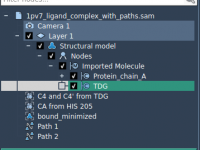
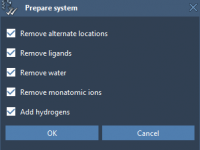
Cleaning Up Protein Structures for Reliable Conformational Interpolation
When modeling conformational transitions between protein states, one of the most overlooked yet crucial steps is structure preparation. Whether you’re setting up umbrella sampling, visualizing intermediate states, or refining with steered molecular dynamics, how you prepare your structures can determine…
Making Room for Clarity: Visual Disassembly of Molecular Structures in SAMSON
Exporting Ligand Trajectories in PDB Format Without the Extra Work
Avoid Repeating Yourself: Add GROMACS Index Groups Once for All Your Simulations
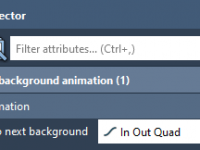
Smooth Transitions for Scientific Storytelling with Background Animations
A Practical Way to Review Your Molecular Trajectories in SAMSON
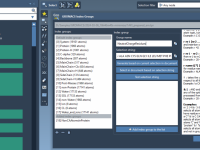
Quickly Find What You Need: Attribute Filtering in the SAMSON Inspector
Faster Nanotube Building with Pattern Editors: A Step-by-Step Guide
Controlling Molecular Visibility in Time with the Hidden Animation
Cleaning Up Protein Structures for Reliable Conformational Interpolation
When modeling conformational transitions between protein states, one of the most overlooked yet crucial steps is structure preparation. Whether you’re setting up umbrella sampling, visualizing intermediate states, or refining with steered molecular dynamics, how you prepare your structures can determine…