





Category: Uncategorized
Molecular Modeling Isn’t Easy—But SAMSON’s Interactive Tutorials Make It Simpler
Working with Molecular Dynamics Trajectories in SAMSON
Molecular modeling and simulation often involve handling large trajectory files produced by molecular dynamics engines like GROMACS, AMBER, NAMD, LAMMPS, and Tinker. However, switching between simulation and visualization tools can feel like constantly battling with incompatible data formats and slow…
Avoiding Costly Mistakes with the History Feature in SAMSON
Bring Your Own Code: Integrate External Tools into SAMSON Easily
Many researchers and developers in molecular modeling rely on custom scripts, external executables, or web services to perform specific tasks—such as docking, calculations, or data analysis. However, managing multiple tools can be time-consuming, especially when interoperability isn’t seamless. This is…
Quickly Select and Show Only What Matters in Your Molecular Scene
How to Quickly Target File Nodes in SAMSON with NSL Filters
Make Molecules Fade, Not Teleport — Disappearing Objects in SAMSON
Tired of Repeating Molecular Simulations? Use Batch Projects in SAMSON
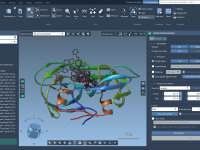
Paths vs. Conformations: Which to Use with the P-NEB Method in SAMSON?
When modeling molecular transitions such as ligand unbinding or conformational changes in biomolecules, predicting realistic pathways is often critical, but far from straightforward. Finding the most efficient and accurate approach can save time and computing resources, especially when working with…
Molecular Modeling Isn’t Easy—But SAMSON’s Interactive Tutorials Make It Simpler
Working with Molecular Dynamics Trajectories in SAMSON
Molecular modeling and simulation often involve handling large trajectory files produced by molecular dynamics engines like GROMACS, AMBER, NAMD, LAMMPS, and Tinker. However, switching between simulation and visualization tools can feel like constantly battling with incompatible data formats and slow…
Avoiding Costly Mistakes with the History Feature in SAMSON
Bring Your Own Code: Integrate External Tools into SAMSON Easily
Many researchers and developers in molecular modeling rely on custom scripts, external executables, or web services to perform specific tasks—such as docking, calculations, or data analysis. However, managing multiple tools can be time-consuming, especially when interoperability isn’t seamless. This is…
Quickly Select and Show Only What Matters in Your Molecular Scene
How to Quickly Target File Nodes in SAMSON with NSL Filters
Make Molecules Fade, Not Teleport — Disappearing Objects in SAMSON
Tired of Repeating Molecular Simulations? Use Batch Projects in SAMSON
Paths vs. Conformations: Which to Use with the P-NEB Method in SAMSON?
When modeling molecular transitions such as ligand unbinding or conformational changes in biomolecules, predicting realistic pathways is often critical, but far from straightforward. Finding the most efficient and accurate approach can save time and computing resources, especially when working with…