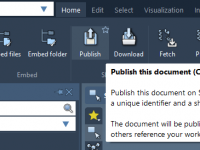
Collaboration lies at the heart of molecular design. Whether you’re part of a research group, teaching a course, or developing computational tools, the ability to share molecular structures and project files seamlessly can save time and improve reproducibility. In SAMSON,…
If you’ve ever created molecular models for presentations or publications, you’ve probably encountered this common frustration: jagged edges that ruin an otherwise beautiful visualization. You’re not alone. One of the frequent complaints among molecular modelers is the aliasing effect—rough or…
Creating molecular animations can be a powerful way to communicate simulations, docking, assembly, and other molecular processes. But what if part of your structure should not move during the animation? For example, maybe you’re modeling how a ligand binds to…
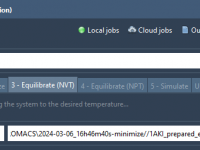
Running molecular dynamics (MD) simulations using GROMACS can be computationally demanding. Whether you’re studying protein folding, drug binding, or materials at the atomic level, the need for high-performance resources often causes significant delays or bottlenecks. This is especially true when…
When working with complex molecular projects in SAMSON that include multiple structural models, segments, and numerous atoms, simple navigation can become time-consuming. If you’ve ever tried to locate specific types of folders—say, those containing more than 1000 atoms or including…
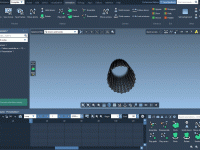
When generating interpolated motion paths between protein conformations, one common frustration is encountering errors due to unclean structures. For example, SAMSON’s ARAP Interpolator may halt with the message: “Cannot proceed because the structure does not make one connected component”. This…
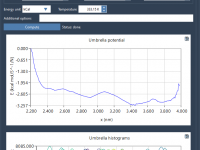
In molecular simulations, the Potential of Mean Force (PMF) is a valuable tool to quantify free energy profiles along a reaction coordinate. Whether you’re studying ligand binding, ion transport, or conformational changes, accurately computing the PMF is crucial to extract…
One of the frequent bottlenecks in setting up molecular dynamics (MD) simulations is choosing and providing the correct input structure. A simple oversight in input file selection can lead to simulation failure or, worse, incorrect results. Fortunately, the GROMACS Wizard…
Creating molecular animations often involves guiding the viewer’s attention to a specific region of interest. However, one common challenge is zooming in without inadvertently shifting the focus point or distorting camera effects like fog or depth-of-field. If you’ve ever struggled…
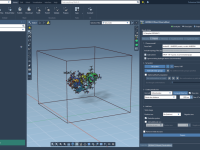
When working with protein structures from the Protein Data Bank (PDB), researchers often encounter a challenge: PDB files typically contain only one asymmetric unit, not the full biological assembly. This is problematic when trying to model or simulate realistic protein…