Streamlining Atom Trajectory Export for Molecular Modeling
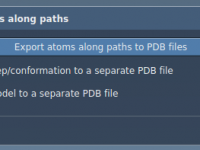
Tracking atom trajectories through molecular pathways forms the backbone of simulations in molecular modeling. However, many researchers face challenges when trying to extract atomic coordinates along defined paths for advanced analyses like reaction coordinate studies or free energy calculations. If…