





When setting up molecular dynamics simulations using GROMACS, one common but often confusing step is defining your system’s boundary conditions. Molecular modelers frequently ask: Which unit cell shape is best for my system?. This blog post explores this issue and explains how GROMACS Wizard in SAMSON helps streamline the choice of unit cell shapes for periodic boundary condition simulations.
Boundary conditions in GROMACS are periodic. That means the system you’re simulating is virtually repeated infinitely in all directions to approximate bulk behavior. But this leads to another challenge—how to pick the most space-efficient and simulation-friendly shape for your unit cell?
Why unit cell shape matters
Your choice of unit cell can have a big impact on simulation performance. If your solute (say a biomolecule) is roughly spherical, placing it in a cubic box wastes space—about 29% more solvent molecules are required compared to more compact shapes. This not only increases simulation time but also CPU usage.
SAMSON’s GROMACS Wizard supports several unit cell shapes:
| Unit cell shape | Representation |
|---|---|
| Cubic |  |
| Orthorhombic |  |
| Triclinic |  |
| Rhombic dodecahedron |  |
| Truncated octahedron |  |
Which one to choose?
The cubic and orthorhombic boxes are straight-forward but less efficient for spherical molecules. For proteins or other globular molecules in solution, the rhombic dodecahedron or the truncated octahedron are recommended. These are better approximations of a sphere, so fewer solvent molecules are required to achieve a given clearance between periodic images.
As an example, a rhombic dodecahedral box has only 71% the volume of a cube with the same image distance. That means nearly 30% savings in solvent molecules—and computational time—for a roughly spherical macromolecule. That’s a meaningful performance gain, especially in large-scale or long-duration simulations.
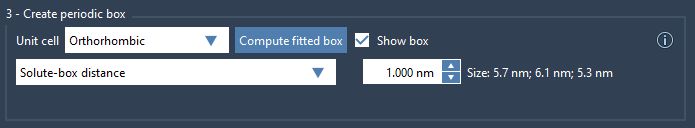
Setting up in GROMACS Wizard
You can choose your unit cell shape during system preparation in GROMACS Wizard. Depending on your project, you have two main options for fitting the box:
- Box lengths: Define the absolute size of the box. Useful if you want uniform box sizes across multiple system conformations (e.g., in a batch project).
- Solute-box distance: Define how far the molecule must be from the box walls (typically at least 1.0 nm). This is more flexible for systems undergoing conformational changes.
Practical tips
- Add at least 1.0 nm between the solute and the box edges to satisfy the minimum image convention.
- When importing trajectories from GROMACS, SAMSON attempts to auto-detect the unit cell shape, but this can be adjusted manually in the importer dialog.
Choosing the right box shape optimizes your simulation’s physical realism and computational cost. For globular systems in solution, consider switching from cubic to rhombic dodecahedral or truncated octahedral next time—it might save time without compromising accuracy.
To learn more, visit the full documentation page: https://documentation.samson-connect.net/tutorials/gromacs-wizard/periodic-boundary-conditions/
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at https://www.samson-connect.net.