Mastering Node Selection with Logical Operators in SAMSON
Boost Your Molecular Modeling: A Dive into SAMSON’s Interactive Simulations
Effortless Python Package Management in SAMSON
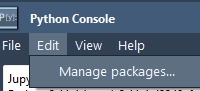
For molecular modelers, workflows often require integrating external Python packages—be it for advanced simulations, custom data analysis, or machine learning implementations. However, installing and managing these packages can sometimes turn into a daunting task. This is where SAMSON’s integrated Python…
Simplifying Protein Preparation for Transition Path Analysis
Simplify Molecular Modeling with Custom File Importers in SAMSON
A Quick Guide to Node Types in SAMSON
Streamlining Polymer Design with Sequence Registration
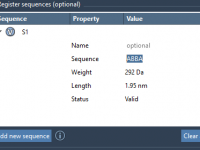
One of the challenges in molecular modeling and materials design is creating complex, custom polymers with repeating sequence patterns. Whether you’re constructing synthetic polymers for material property predictions or drug-polymer conjugates, manually assembling these structures can be tedious and error-prone.…