





Mastering Custom Molecular Dynamics Parameters in GROMACS Wizard
Essential Steps to Prepare Your Protein Model for Transition Pathway Searches
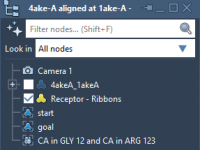
Effortless Protein Preparation: Saving Time Before Docking
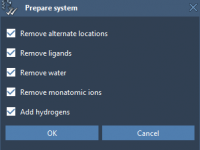
Protein docking simulations often require extensive preprocessing to ensure accurate and reliable results. For molecular modelers, preparing proteins can sometimes feel tedious and error-prone, especially when managing large datasets or dealing with specific structural inconsistencies. Did you know that the…
Getting Started with SAMSON: A Handy Guide for First-Time Users
Understanding Segment Attributes for Molecular Modeling in SAMSON
When working on integrative molecular design, molecular modelers often face challenges in efficiently organizing and analyzing their molecular structures. SAMSON’s Node Specification Language (NSL) offers powerful tools for navigating such challenges by enabling precise and flexible querying of structural attributes.…
Demystifying Property Model Attributes for Molecular Modelers
Molecular modeling often requires precise definitions and manipulations of structural and functional properties to ensure accurate simulations or visualizations. For those diving into SAMSON’s Node Specification Language (NSL), the propertyModel attribute space offers a powerful way to define and explore…
Simplify Molecular Modeling: Customizing SAMSON’s Interface with Ease
For molecular modelers, an intuitive and personalized workflow is crucial. Yet, many face challenges aligning their software interface with their unique preferences. Fortunately, SAMSON, the integrative molecular design platform, allows users to fully customize its interface for a streamlined experience.…
Ensuring Seamless Compatibility of SAMSON Extensions.
Understanding Graphics Requirements for SAMSON
As a molecular modeler, ensuring a seamless experience with your tools is essential. SAMSON, the powerful molecular design platform, relies on specific graphics capabilities to provide accurate simulations and visualizations. Misconfigured hardware or operating in environments lacking proper graphical support…