Streamlining Molecular Modeling with Symmetry Detection in SAMSON.
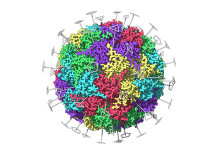
For molecular modelers working with large protein complexes, viral capsids, or other biological assemblies, detecting and understanding symmetry is both a critical need and a time-saving trick. SAMSON offers a dedicated Symmetry Detection extension that simplifies this process, allowing researchers…
Streamlining Molecular Dynamics with GROMACS Wizard: Batch Computations
Refining Molecular Geometry: A Practical Guide to the FIRE Minimizer in SAMSON

Molecular modelers often grapple with the challenge of finding stable and realistic structures that align with energy minima. This is where geometry optimization becomes essential. Without refined molecular geometries, simulations risk becoming inefficient or inaccurate. A common bottleneck arises with…
Unlocking the Power of Per-Attribute Color Schemes in Molecular Models
Effortlessly Capture Stunning Molecular Viewports in SAMSON
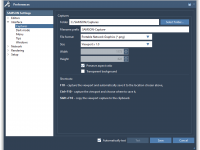
Unlocking the Versatility of SAMSON’s Supported File Formats
Practical Tips for Visualizing Specific Atoms in Molecular Structures

When working on molecular modeling, selecting and visualizing atoms corresponding to specific criteria can be a recurring challenge. Whether you’re analyzing specific molecular fragments, modifying atomic properties, or fine-tuning nanoscale designs, having a systematic approach to selecting atoms is essential.…
Streamlining Molecular Dynamics with Cloud Jobs in GROMACS Wizard
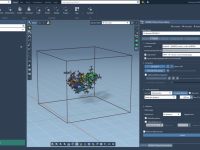
Molecular dynamics simulations often demand significant computational power, especially when dealing with large systems or batch computations. For researchers and molecular modelers, the ability to perform such heavy calculations without relying solely on local hardware can be game-changing. This is…