Understanding CRYST1 and BIOMT Symmetry in Protein Structures
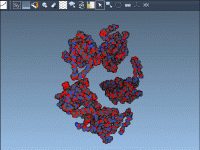
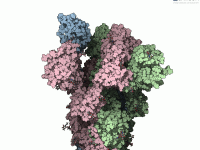
Have you ever wondered how proteins arrange themselves in their crystal structures or biological assemblies? For molecular modelers, reconstructing these arrangements is crucial for understanding protein interactions and designing experiments. The Symmetry Mate Editor in SAMSON offers a powerful way…