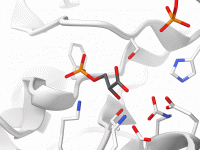
Easily Align Protein Sequences and Structures Using SAMSON’s Protein Aligner
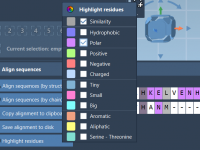
Aligning protein sequences and structures is critical for molecular modelers aiming to understand functional similarities, evolutionary relationships, or to design novel molecules. However, the process can be time-consuming, especially when working with multiple chains or complex models. Fortunately, SAMSON’s Protein…