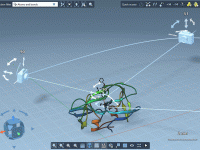
Transform Reaction Pathways into Precise Umbrella Sampling Windows
Demystifying Property Model Attributes in Molecular Design
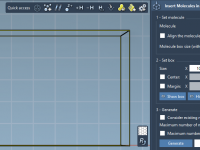
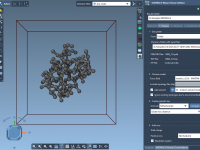
Mastering the Preparation of Coarse-Grained Systems with SAMSON’s GROMACS Wizard
Understanding the Property Model Attributes in SAMSON
Effortlessly Select Nodes with SAMSON’s Node Specification Language (NSL)
Effortlessly Track Molecular Movements with ‘Follow Atoms’
Understanding and Querying Residue Charges in Molecular Models.
As a molecular modeler, understanding the charge states of residues is essential for tasks such as analyzing protein structures, predicting molecular interactions, and studying electrostatic properties. The residue.charge attribute in SAMSON’s Node Specification Language (NSL) provides a powerful way to…
Streamlining Protein Visualization with SAMSON Visual Models

For molecular modelers, accurately visualizing complex structures like proteins can often feel overwhelming, especially when dealing with large systems or seeking specific graphical representations to highlight structural details. SAMSON, the integrative molecular design platform, offers a comprehensive solution through its…