





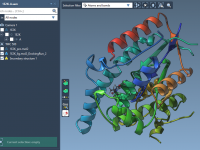
Effortlessly Visualize Molecular Trajectories in Reverse
Simplifying Molecular Modeling with SAMSON Extensions

Mastering Structural Group Attributes in SAMSON for Molecular Modeling
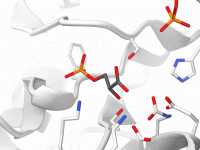
Streamline Your Molecular Visualizations with Visual Presets
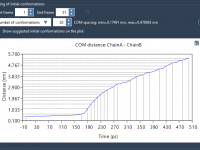
Understanding Conformation Attributes in SAMSON’s Node Specification Language.
Molecular modeling often involves the need to filter or analyze conformations based on specific attributes. Whether you’re optimizing structures or comparing molecular conformations, being able to define clear attribute-based filters is crucial. Within SAMSON’s Node Specification Language (NSL), the conformation…
Integrating All Your Research into One Place
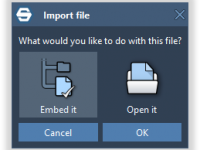
Understanding Crystalline Defects in Diamonds with SAMSON
Simplifying Molecular Design Tasks with SAMSON AI’s /script Command
Mastering the Preferences Panel in SAMSON for Efficient Molecular Modeling.
For molecular modelers, optimizing your workspace to suit your modeling objectives is critical. Customizing software settings can make workflows smoother, more predictable, and efficient. SAMSON’s Preferences panel aims to tackle this very need, providing granular control over interface behavior, appearance,…