Design Lipid Layers Around Proteins with Ease
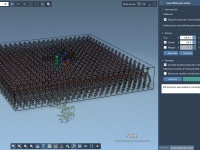
Molecular modelers often face the challenge of creating realistic lipid environments around proteins for their simulations. Whether you’re preparing for molecular dynamics or want to understand interactions in membrane-like environments, constructing these systems can often be time-consuming and error-prone. The…