Streamlining Molecular Design Workflows with SAMSON Apps.
For molecular modelers, finding efficient ways to execute complex workflows, integrate external tools, and expand software functionality is a recurring challenge. Enter SAMSON Apps—powerful additions designed to enhance the versatility of the SAMSON molecular design platform. Whether you are connecting…
Mastering Structural Model Attributes in SAMSON
How to Bring Molecular Models to Life with SAMSON’s Color Schemes.
Mastering Path Attributes in SAMSON’s Node Specification Language
Unlocking Animation Effects in Molecular Modeling
Mastering Static Views with the Hold Camera Animation in SAMSON
No Administrator? No Problem: Install SAMSON Without Admin Rights.
Tracking Solvent-Accessible Surface Area for Your Molecular Studies
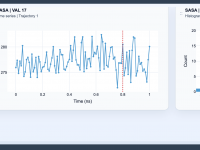
Molecular modelers often grapple with understanding how different molecular components interact with their environments, such as how solvent-exposed areas change during processes like binding, folding, or conformational shifts. Have you been looking for a straightforward way to quantify exposure, compaction,…