Mastering Attribute Inspection in SAMSON’s Inspector Tool
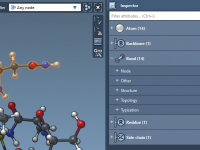
Streamline Molecular Design with Quick Groups in SAMSON.
Molecular modeling can be a highly complex task, especially when dealing with intricate systems like large biomolecules. Navigating between different regions of interest, applying specific transformations, or comparing structural elements often adds to this complexity. To address this, SAMSON introduces…
Simplified Steps to Install SAMSON and Get Started
Effortless Molecular Rearrangement with SAMSON’s Undock Animation
Molecular modelers frequently face challenges when it comes to manipulating complex structures, especially repositioning atoms or groups during presentations or simulations. Rearranging molecular components manually is time-consuming, error-prone, and often disrupts structural integrity, which can hinder research workflows and effective…
Master Precise Manipulation with Move Editors in SAMSON
Mastering Light Attributes in SAMSON’s Node Specification Language (NSL)
Simplify Molecular Modeling with Conformation Attributes in SAMSON
Molecular modelers often spend significant time navigating complex datasets to identify specific conformations for their structures. Filtering through extensive molecular data to locate, for instance, conformations with a certain number of atoms or specific selection criteria can be daunting without…