





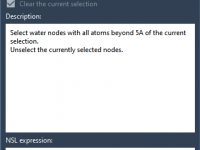
Efficiently Removing Unnecessary Water Molecules in Your Molecular Simulations
Mastering the Dolly Camera Effect for Molecular Animations
For molecular modelers, visualizing molecular systems with precision and fluidity is essential, whether you’re preparing a presentation or analyzing complex structures. Achieving a professional-grade animation often involves creating realistic motion effects, and that’s where the Dolly Camera animation in SAMSON…
Streamline Molecular Motion Analysis with Rotate Animation in SAMSON
One of the common challenges in molecular modeling is effectively visualizing and analyzing the motion of molecular groups. Understanding these motions is crucial for exploring molecular interactions, designing new materials, or discovering drugs. SAMSON, the integrative molecular design platform, offers…
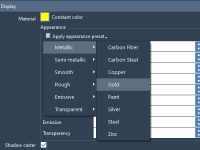
Mastering Material Control for Molecular Models.
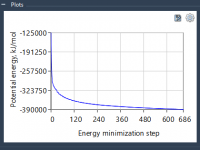
Simplifying Molecular Geometry Optimization with the FIRE Minimizer

Molecular modelers often face a critical challenge: preparing stable and realistic molecular geometries for simulations or molecular design workflows. Whether you’re cleaning up a structure before running simulations or optimizing geometries during interactive modeling, time and accuracy matter. This is…
Streamlining Energy Minimization with SAMSON’s GROMACS Wizard
How to Minimize Only Part of a Molecule in SAMSON
Understanding Conformation Attributes for Molecular Modeling
When working on molecular modeling, one common challenge is effectively analyzing and managing molecular conformations to extract useful insights. SAMSON, with its powerful integrative molecular design platform, offers tools to address this challenge efficiently. One particularly useful system within SAMSON…