Streamline Molecular Design with Positional Analogue Scanning in SAMSON
Molecular modeling often requires exploring how small changes in a molecule’s structure affect its properties, like binding affinity or interactions with target proteins. However, manually generating molecular analogs for such studies can be tedious and time-consuming. Enter positional analogue scanning,…
Understanding Atom Geometries in Molecular Modeling.
If you’re a molecular modeler, you’ve likely encountered situations where defining atom geometry precisely is critical. Whether crafting small molecules or designing complex protein-ligand interactions, atom geometry can play a significant role in capturing molecule behavior and predicting outcomes. SAMSON’s…
Mastering Vertical Camera Movement with the Pedestal Camera Animation
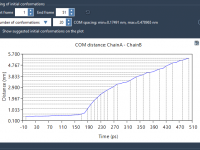
Streamline Molecular Dynamics with GROMACS in the Cloud
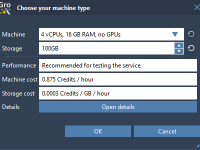
For molecular modelers, one of the biggest challenges can be the need for intensive computational resources. Handling large molecular systems or running long dynamics simulations can overburden local machines, making it difficult to complete projects efficiently. Fortunately, SAMSON’s GROMACS Wizard…
Simplifying Molecular Modeling: Preparing Coarse-Grained Systems with GROMACS Wizard
The Importance of Protein Validation for Molecular Modeling Success
Enhancing Molecular Depth with Ambient Occlusion in SAMSON

When dealing with complex molecular visualizations as a molecular modeler, one significant challenge is achieving clear depth perception. Understanding the spatial relationships between molecular regions is crucial for presentations, research, or educational purposes. In this blog post, we’ll explore how…