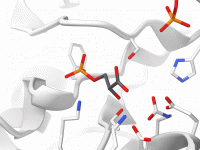
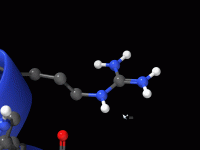
Streamlining Molecular Modeling with File Attributes in SAMSON
Molecular modelers often deal with complex data structures, requiring precise organization and customization to efficiently manage their projects. Whether it’s targeting specific molecular files or managing node-specific details, SAMSON provides powerful tools to simplify these tasks, including its file attributes.…