





Effortless Document Sharing for Molecular Modeling Projects
Effortless Transition Paths for Protein Conformations with ARAP Interpolation
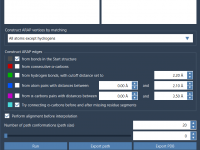
Protein modeling often involves analyzing structural transitions between conformations. These transitions are critical for understanding biochemical mechanisms, simulating reaction pathways, or setting up free energy simulations. However, generating smooth and biologically meaningful transition paths can be a challenge. This is…
Understanding Structural Group Attributes in SAMSON’s Node Specification Language
A Smooth Introduction to the Appear Animation in SAMSON
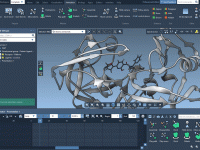
Refining Molecular Dynamics in GROMACS Wizard: Custom Parameters Made Easy
Streamlining Teamwork in Molecular Design with SAMSON Connect
Exploring Molecule Attributes in SAMSON’s Node Specification Language
Streamline Molecular Modeling with Your Custom GROMACS Installation
Refining Molecular Presentations with the Stop Animation in SAMSON
Molecular presentations play a crucial role in conveying complex structural insights, whether you are presenting your findings to colleagues, teaching students, or preparing professional reports. However, one common challenge that molecular modelers face is creating clear, segmented animations that allow…