Preview and Generate Protein Symmetry Mates in Real Time
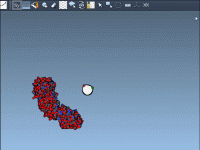
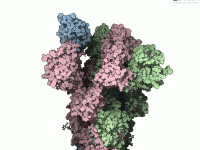
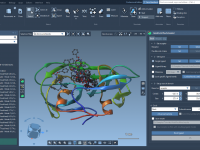
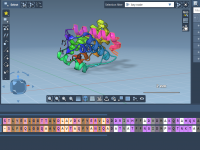
Exploring the arrangement of protein subunits is essential in structural biology, particularly when transitioning from crystallographic units to biologically relevant assemblies. A recurring challenge for molecular modelers is determining how individual protein chains relate to their symmetric counterparts, especially when…