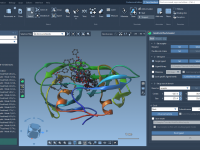
Making Molecular Documents Self-Contained with Universal File Embedding in SAMSON
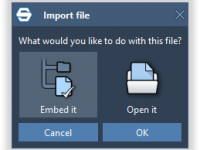
One common pain point for molecular modelers is managing the many files associated with a project—molecular structures, Python scripts, input/output files, figures, research papers, and other resources. These files often get scattered across folders, complicating versioning, backups, collaboration, and reproducibility.…
Creating and Breaking Bonds in Real Time with IM-UFF
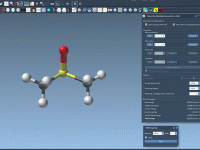
One common challenge for molecular modelers is editing molecular structures while maintaining physical realism. Whether you’re designing molecules, studying reaction pathways, or exploring hypothetical structures, you often need to dynamically add or break bonds. Doing this manually can be time-consuming…