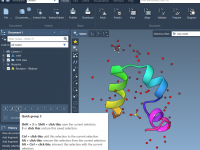
Streamline Molecular Selections with Quick Groups in SAMSON
Why Your SAMSON Extension May Not Load (and How to Fix It)
Connecting External Tools to SAMSON Without Rewriting Code
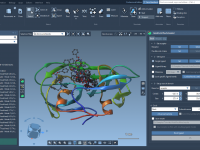
From Flat to Focused: How Ambient Occlusion Enhances Molecular Visualization

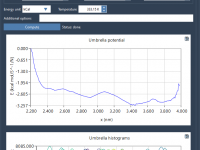
Wrapping a Protein in Lipids: A Practical Guide
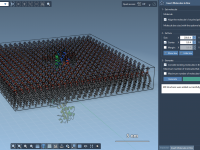
For molecular biophysicists and structural biologists, preparing lipid bilayers around membrane proteins can be an essential yet tedious task. Whether you’re setting up simulations of transmembrane channels, studying membrane-protein interactions, or exploring lipid dynamics, aligning lipids precisely around your protein…