Achieve Precision with Constrained Simulations in SAMSON
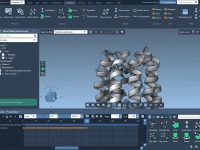
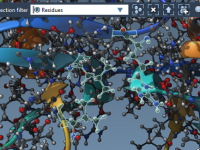
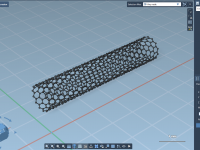
Molecular modeling often requires a delicate balance between realism, precision, and computational efficiency. When simulating complex nanosystems, one frequent challenge is controlling atom positions to mimic real-world constraints while performing simulations. This article introduces a valuable method in SAMSON to…