Streamlining Molecular Visualizations with Animation Effects in SAMSON
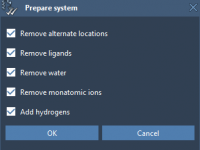
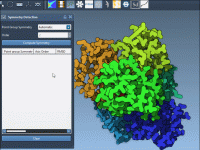
Visualizing molecular processes effectively is a vital challenge faced by many molecular modelers. Whether you’re creating presentations for your peers, preparing educational material, or simply exploring complex molecular dynamics, adding animations can significantly enhance clarity and engagement. With SAMSON, the…