





Author: OneAngstrom
Streamline Vertical Camera Movements with the Pedestal Camera Animation.
Molecular modeling often demands precise control over the viewpoint, especially when exploring structures closely or presenting data effectively. Adjusting a camera's position within a three-dimensional molecular scene is time-intensive and can interrupt the modeling workflow. This is where the Pedestal…
Leveraging Note Attributes in SAMSON for Efficient Molecular Modeling
Streamlining Molecular Modeling with Structural Group Attributes
Boost Efficiency with SAMSON’s Advanced Selection Tools
Mastering Ligand Flexibility for Realistic Docking Simulations
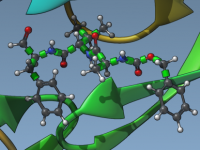
For molecular modelers striving to improve the accuracy and realism of protein-ligand docking, accounting for ligand flexibility is crucial. Flexibility allows the exploration of different conformations and interactions within the binding site, making docking outcomes more reliable. However, controlling ligand…
Effortlessly Track Atoms with SAMSON’s Follow Atoms Animation
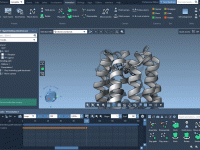
Simplify Molecular Visualization with Pulse Animation in SAMSON
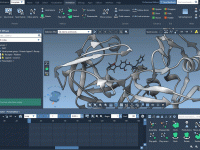
Mastering Camera Perspectives for Molecular Animations
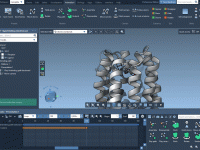
Streamline Molecular Animations with Truck Camera
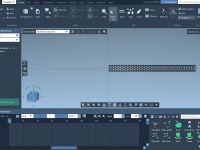
Molecular modelers working on dynamic presentations or analyses often face a common challenge: creating smooth, horizontal camera transitions that effectively showcase their systems. A well-positioned camera animation can make all the difference in presenting complex molecular structures and interactions in…