Author: OneAngstrom
Effortlessly Expand Your Molecular Modeling Toolbox with SAMSON Extensions

For molecular modelers, one persistent challenge is finding versatile and specialized tools that cater to unique scientific needs. Imagine if you could easily integrate functionalities such as advanced simulation algorithms, specialized visualization tools, or import/export functionalities without switching platforms. SAMSON…
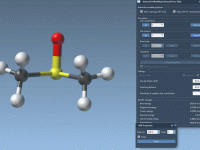
Understanding Visual Model Attributes in SAMSON
Mastering Extensions in SAMSON: Simplify Molecular Modeling with Ease
Simplifying Molecular Modeling Workflows with Python in SAMSON.
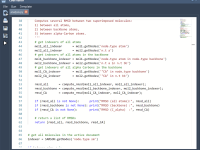
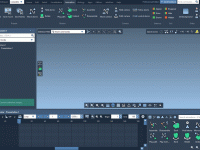
Creating Seamless Transitions with Background Animations in SAMSON.
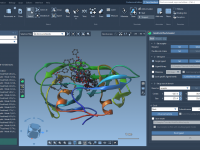
Streamlining Molecular Visualizations: Applying Visual Presets in SAMSON
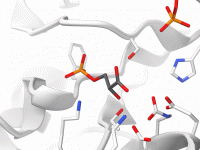
For molecular modelers, especially those working with complex molecular systems like proteins and ligands, visualizing structures effectively and efficiently can be a significant challenge. How do you represent intricate details without spending hours configuring visual parameters? That’s where visual presets…
Step-by-Step Guide to Building Carbon Nanotube Models

Carbon nanotubes (CNTs) are essential in nanotechnology, molecular design, and advanced material science. Their unique properties make them invaluable for designing nanodevices, sensors, and membranes, as well as exploring electronic and mechanical behaviors. However, creating precise CNT models can be…